编辑 | 萝卜皮
宏基因组组装基因组 (MAG) 为利用宏基因组测序数据探索微生物「暗物质」提供了宝贵的见解。
然而,人们越来越担心 MAG 中的污染可能会严重影响下游分析的结果。目前的 MAG 净化工具主要依赖于标记基因,并没有充分利用基因组序列的背景信息。
为了克服这一限制,香港浸会大学(Hong Kong Baptist University)和英伟达 AI 技术中心(NVIDIA AI Technology Center)的研究人员提出了 Deepurify 来进行 MAG 净化。
Deepurify 使用多模态深度语言模型和对比学习来匹配微生物基因组序列与其分类谱系。它将 MAG 内的 contig 分配给 MAG 分离树,并应用树遍历算法将 MAG 划分为子 MAG,目的是最大化高质量和中质量子 MAG 的数量。
Deepurify 在模拟数据、CAMI 数据集和复杂程度各异的真实数据集上的表现均优于竞品(MDMclearer 和 MAGpurify)。在土壤、海洋、植物、淡水和人类粪便宏基因组测序数据集中,Deepurify 分别使高质量 MAG 的数量增加了 20.0%、45.1%、45.5%、33.8% 和 28.5%。
该研究以「A multi-modal deep language model for contaminant removal from metagenome-assembled genomes」为题,于 2024 年 10 月 7 日发布在《Nature Machine Intelligence》。

利用短读宏基因组测序数据进行基因组组装已成为研究复杂环境中微生物暗物质的常用方法。然而,单个重叠群仅捕获完整微生物基因组的一个片段。因此,需要进行重叠群分箱,将具有相似序列特征和丰度的重叠群分组以代表微生物基因组。
有研究表明,MAG 污染是宏基因组组装中重叠群分箱过程中的一大挑战。已经开发出 MAGpurify 和 MDMcleaner 等工具来解决此问题,方法是从 MAG 中去除受污染的重叠群。
然而,这些工具有一些局限性。如果核心基因组和受污染基因组的 LCA 属于同一科或属,它们很难区分重叠群。
同时,还有来自源基因组的重叠群的挑战,这些重叠群在参考数据库中是不存在的。此外,这些工具主要关注基因,而忽略了基因顺序和基因组重排等基因组变异。
在最新的研究中,香港浸会大学和英伟达 AI 技术中心的研究人员开发了 Deepurify,一种多模态深度语言模型,用于高分辨率和广义的 MAG 净化。
在训练过程中,Deepurify 使用两个编码器,GseqFormer 和长短期记忆 (LSTM),分别生成基因组序列及其源基因组分类谱系的嵌入。然后,这些嵌入用于对比学习,以建立这两种类型的模态之间的关系。
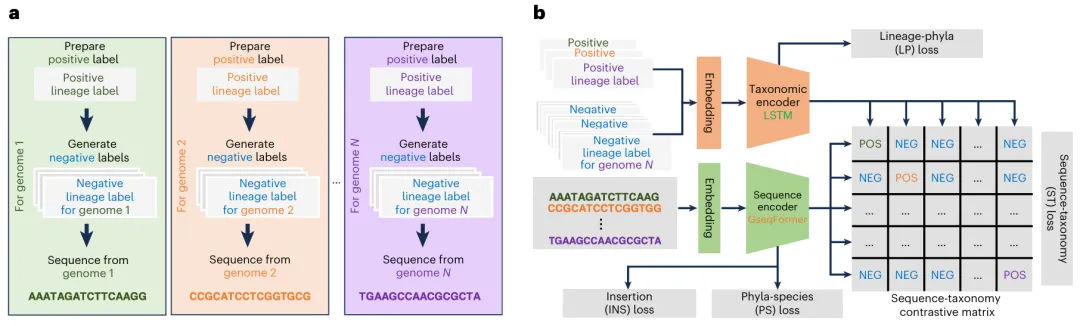
图示:Deepurify 训练流程。(来源:论文)
在净化过程中,Deepurify 首先根据预测的分类谱系评估 MAG 中重叠群的分类相似性。预测的分类谱系用于构建 MAG 分离树,其中每个节点包含特定分类群中具有相同分类谱系的重叠群。每个节点的重叠群根据其序列嵌入和注释的单拷贝基因 (SCG) 分组为子 MAG。
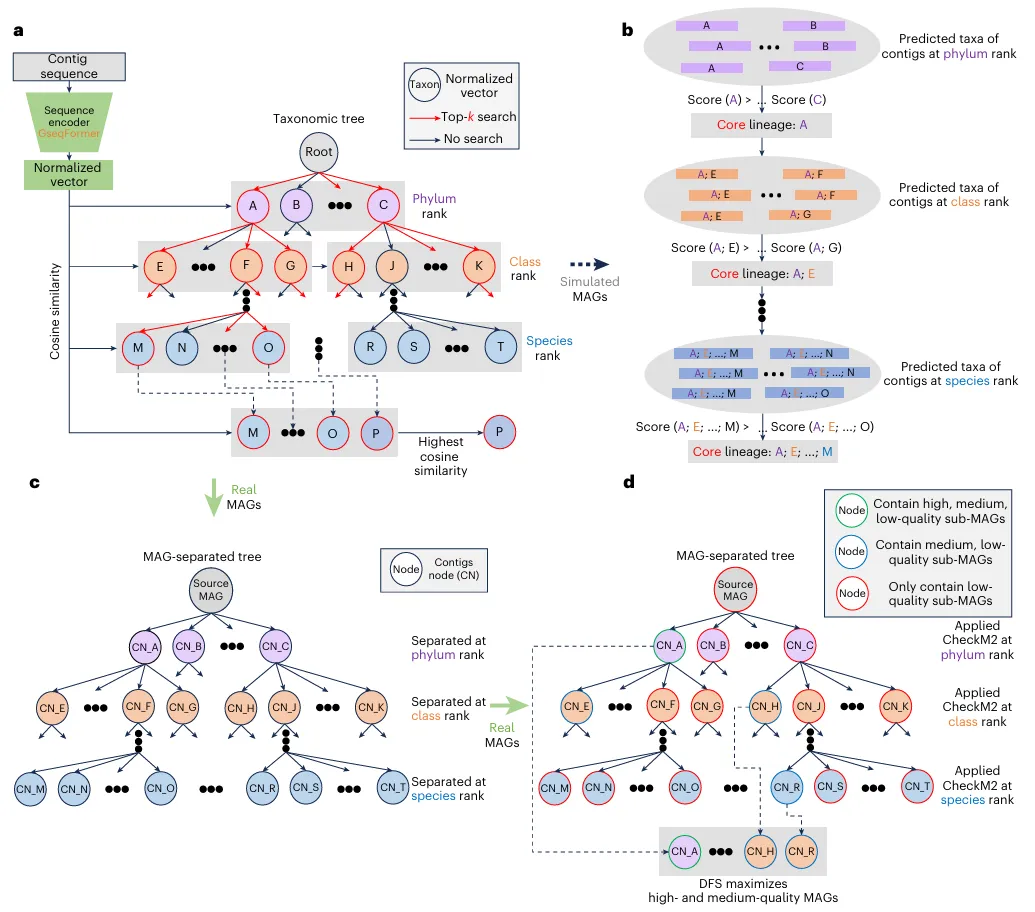
图示:Deepurify 用于 MAG 净化的工作流程。(来源:论文)
研究人员使用了一种树遍历算法来选择子 MAG,旨在从树中最大化高质量和中等质量 MAG 的总数。此外,他们实施了一种名为 Deepurify_Iter 的迭代净化策略,以促进来自多个分箱工具的 MAG 的逐步净化。
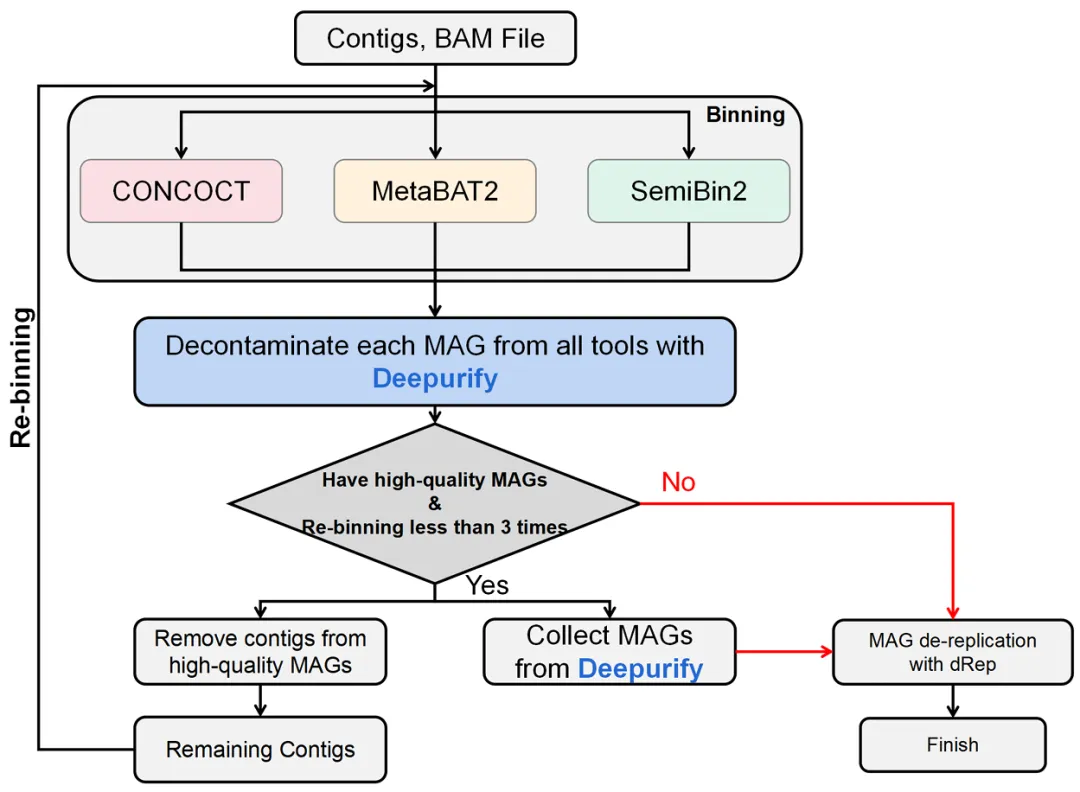
图示:Deepurify_Iter 中使用的迭代净化策略的工作流程。(来源:论文)
对于模拟数据,Deepurify 在 MAG 净化方面的表现优于两种最先进的工具 MAGpurify 和 MDMcleaner,如下图所示。
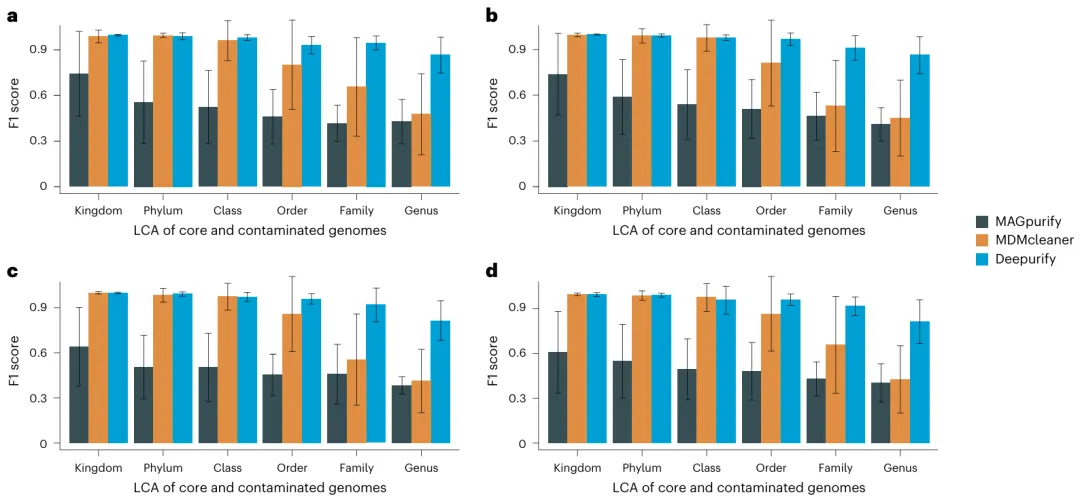
图示:不同污染率下平衡宏观 F1 得分的平均数。(来源:论文)
Deepurify 还展示了出色的泛化能力,即使训练集中没有源基因组,也能准确识别受污染的重叠群,如下图。对于宏基因组解释的关键评估 (CAMI) I 和真实世界的宏基因组测序数据集,研究团队使用 GUNC 来评估净化后的 MAG 的污染水平。
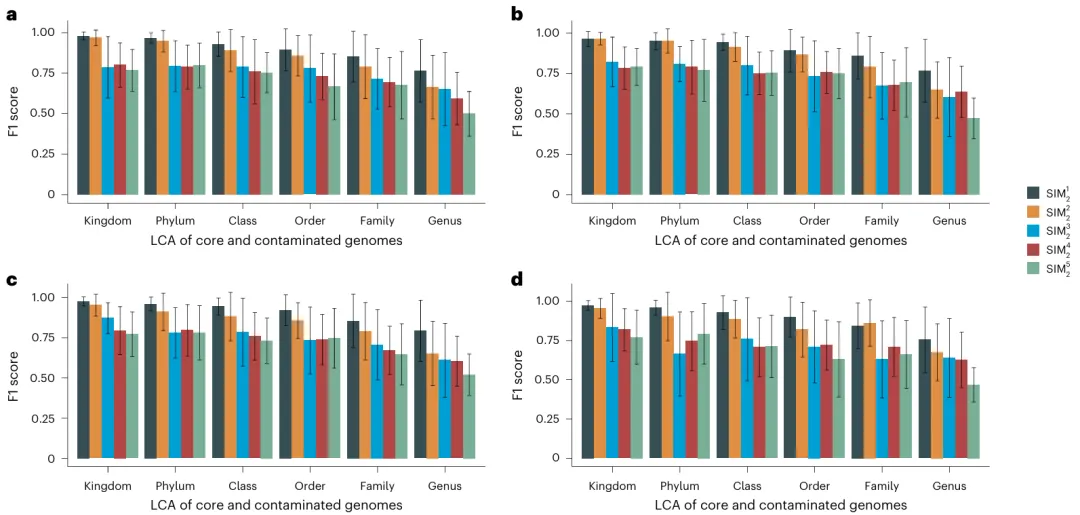
图示:不同污染率下平衡宏观 F1 得分的平均数(训练集中没有源基因组)。(来源:论文)
对于 CAMI I 数据集,研究人员将 MAGpurify、MDMcleaner、Deepurify 和 Deepurify_Iter 应用于由三个 contig 分箱工具生成的 MAG:CONCOCT、MetaBAT2 和 SemiBin2。
结果表明,Deepurify 和 Deepurify_Iter 显著减少了 MAG 的污染,在所有分箱工具中都超过了 MAGpurify 和 MDMcleaner,如下图所示。
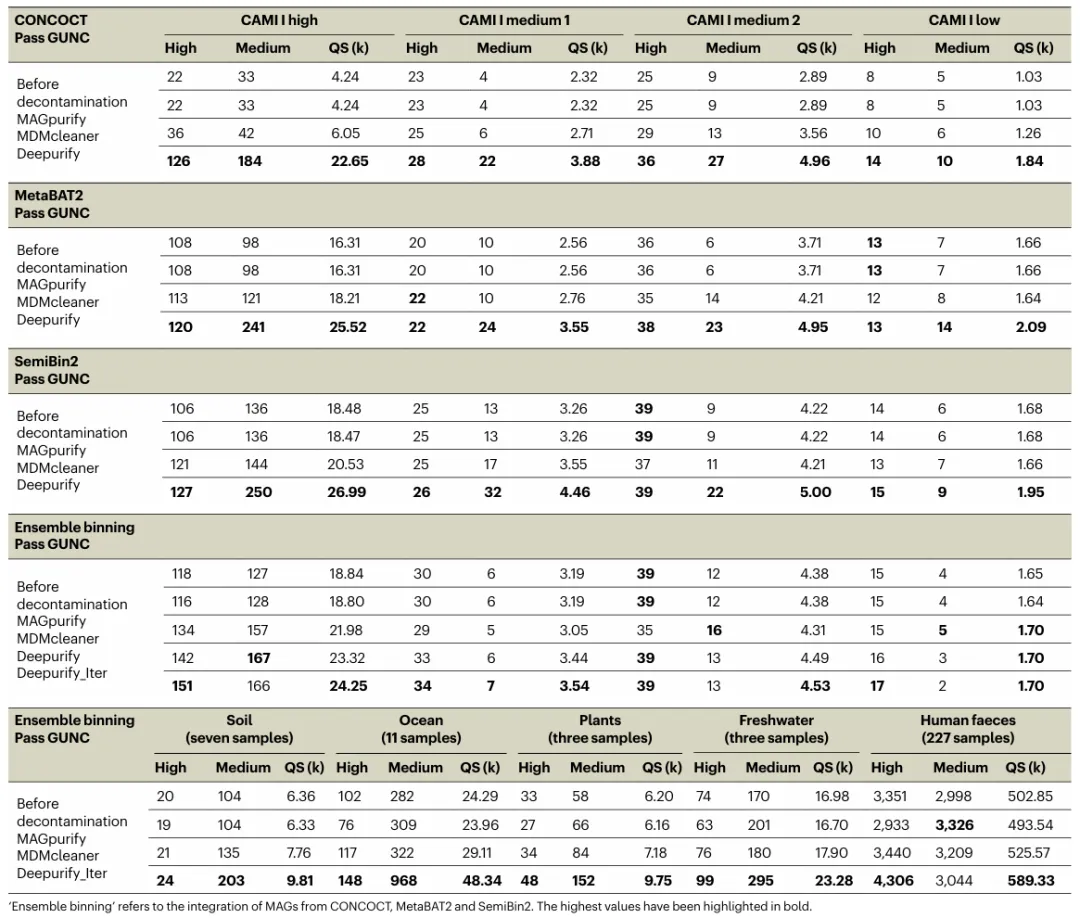
图示:通过 GUNC 污染标准的高质量和中等质量 MAG 的数量,以及 CAMI I 和五个真实世界数据集的质量得分(1k = 1,000)。(来源:论文)
同时,他们将 Deepurify_Iter 应用于现实世界中复杂程度各异的宏基因组测序数据集,包括来自土壤、海洋、植物、淡水和人类粪便的样本。研究结果表明,Deepurify_Iter 显著提高了所有这些样本中 MAG 的质量,高质量 MAG 分别增加了 20.0%、45.1%、45.5%、33.8% 和 28.5%。并且,即使处理来自高度复杂生态系统的 MAG,Deepurify_Iter 的性能仍然保持稳健。
总之,该团队证明了 Deepurify 在从短读长宏基因组组装中去除 MAG 方面具有显著的效果。
论文链接:https://www.nature.com/articles/s42256-024-00908-5