
编辑 | 2049
在量子化学计算中,精确预测分子电子结构一直是一个重要而富有挑战性的课题。
传统的密度泛函理论(DFT)方法虽然计算速度快,但精度有限;而高精度的耦合簇(CCSD(T))方法虽然被视为「金标准」,但其计算成本随分子大小呈指数级增长,难以应用于复杂体系。
最近,麻省理工学院的研究团队开发出一种创新的多任务学习方法,成功将机器学习与量子化学计算相结合,实现了接近CCSD(T)精度的分子电子结构预测。
该研究以「Approaching coupled-cluster accuracy for molecular electronic structures with multi-task learning」为题,于 2024 年 12 月 27 日发布在《Nature Computational Science》。

研究背景
在现代计算化学领域,预测分子电子结构的主流方法是密度泛函理论。作为一种平均场理论,DFT 的系统误差通常是化学精度(1 kcal/mol)的数倍。
近年来,机器学习方法被广泛应用于提高 DFT 计算的精度,但由于这些模型都是基于 DFT 数据训练的,其精度始终无法超越 DFT 本身的理论极限。
另一方面,基于耦合簇理论的 CCSD(T)方法虽然能提供极高的计算精度,但其计算复杂度随电子数的增长呈现 N⁷ 级别的标度,这导致它只能处理包含数百个电子的小分子体系。
研究团队敏锐地发现,如果能将机器学习与 CCSD(T)方法有机结合,可能突破这一技术瓶颈。
MEHnet:融合物理洞察的深度学习框架
研究团队开发的多任务电子哈密顿网络(Multi-task Electronic Hamiltonian Network, MEHnet)采用了独特的物理启发式设计。
该方法首先使用 DFT 获得初始的平均场哈密顿量,作为快速但精度较低的起点。然后通过神经网络预测非局域交换关联修正项,这一修正项能捕捉到电子间的量子关联效应,最终得到接近 CCSD(T)精度的有效单体哈密顿量。
MEHnet 的核心创新在于其多任务学习策略。不同于传统方法仅关注分子能量的预测,MEHnet 同时预测多个物理量,包括偶极矩、四极矩、原子电荷和键级等。这些物理量都源于同一个电子结构表示,通过多任务学习可以相互促进,提高模型的泛化能力。
在技术实现上,MEHnet 采用了 E3-等变神经网络框架(E3-equivariant Neural Network),确保预测结果满足物理系统的旋转不变性。
如图 1 所示,整个计算流程包括输入层、卷积层和输出层三个主要部分。输入层将原子构型编码为图结构;卷积层通过 E3-等变神经网络提取特征;输出层则产生多个量子化学性质的预测值。
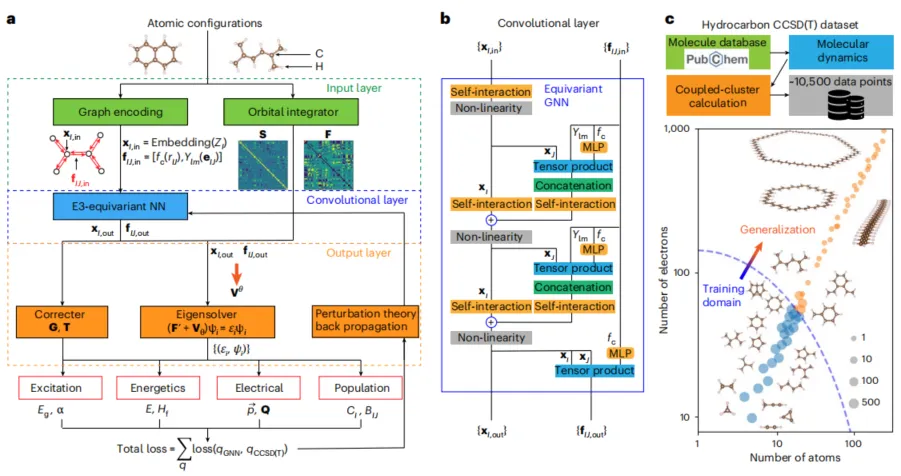
图 1:MEHnet 的计算流程示意图。(来源:论文)
性能评估与实验验证
研究团队在碳氢化合物数据集上对 MEHnet 进行了全面评估。实验设计包括两个维度:
一是通过改变训练集大小(从 10 到 7,440 个原子构型)评估模型的学习效率和泛化能力;
二是与主流的 DFT 方法和机器学习模型进行性能对比。评估指标包括能量预测误差、偶极矩、四极矩、原子电荷等多个物理量的均方根误差。
实验结果表明,MEHnet 在计算效率和预测精度两个方面都实现了突破性进展。
如图 2 所示,MEHnet 的计算成本随分子大小呈近似线性增长(~N¹),而 CCSD(T)方法在理论上渐近标度为 N⁷,计算效率提升约百万倍。
在预测精度方面,对于能量预测,MEHnet 实现了约 0.1 kcal/mol 每原子的误差水平,接近化学精度;对于其他物理量的预测,也普遍优于 B3LYP 等广泛使用的混合泛函方法。
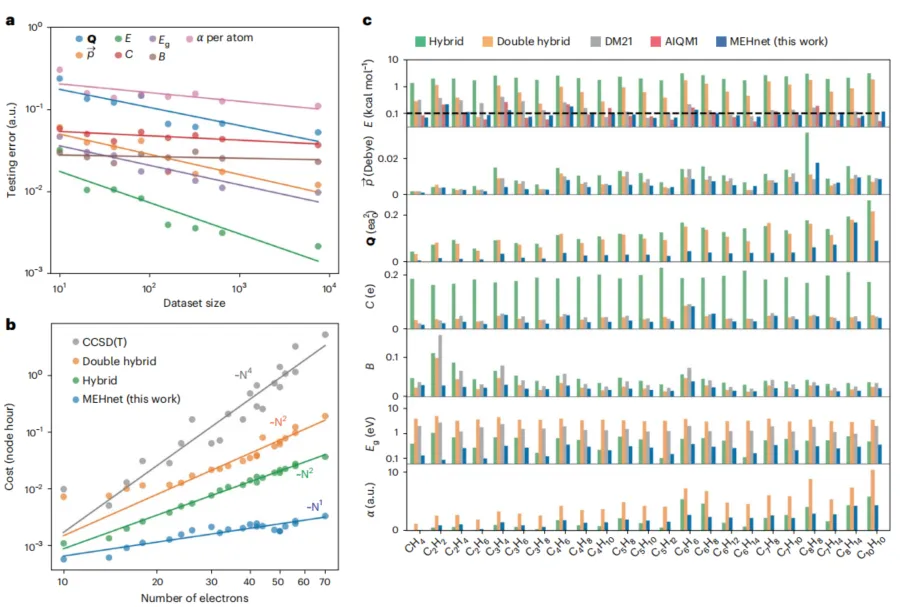
图 2:测试数据集上的模型性能基准。(来源:论文)
结语
MEHnet 的创新在于其能够以 CCSD(T)级别的精度预测分子电子结构,同时保持 DFT 级别的计算效率。通过多任务学习和 E3-等变神经网络的结合,MEHnet 展示了机器学习在复杂系统电子结构预测中的巨大潜力。
这一研究不仅解决了当前量子化学计算中的关键瓶颈,还为未来的跨学科研究提供了新的思路和方法。
论文链接:https://www.nature.com/articles/s43588-024-00747-9


