编辑 | ScienceAI
小分子力场是药物发现中的重要工具,在计算机辅助药物设计中发挥关键作用。化学空间覆盖广泛且高效精确的小分子力场将为药物发现奠定可靠的基础。
尽管基于机器学习的 MLFF(如 ANI-2x,MACE-OFF23 等)能够提供非常精确的小分子势能面预测,但它们的训练需要海量数据量,且推理速度较慢,还存在外推场景不确定度大等问题。这些问题限制了它们在药物发现场景的应用。
以 Espaloma 为代表的一系列工作,在保留传统力场势函数形式的基础上,使用 GNN 预测传统力场参数,一定程度上平衡了力场的精度与效率,为传统力场的发展开创了新的道路。
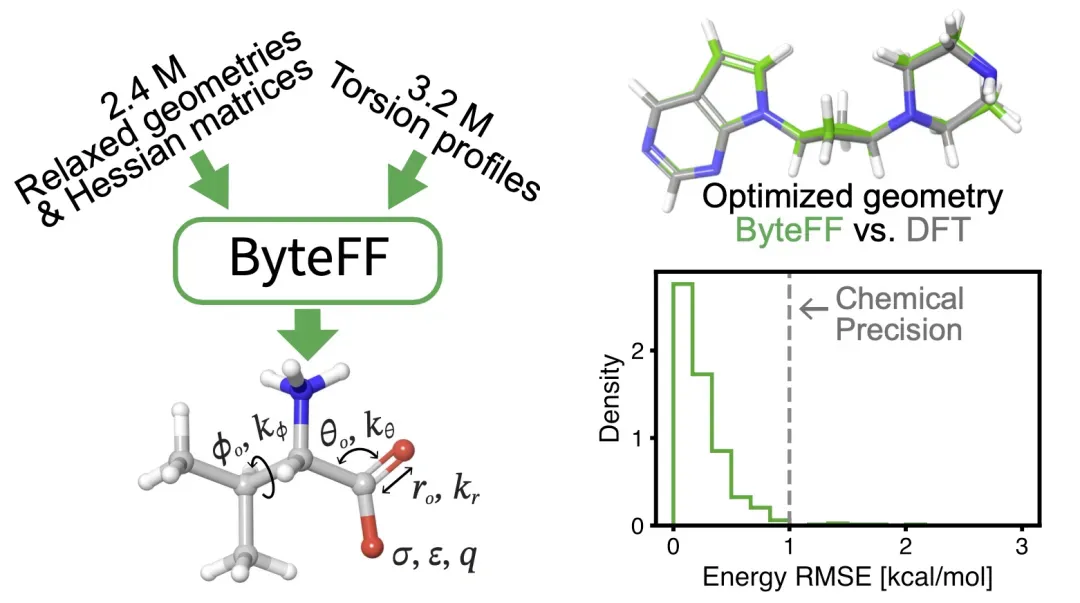
在此基础上,字节跳动的研究人员设计了符合物理约束的模型结构,构建了覆盖广阔化学空间的量化数据集,使用配套的训练方案,开发了 ByteFF 力场。测试结果表明,ByteFF 在结构优化,分子势能面预测等多项指标上都达到了 SOTA 水平。
相关研究以「Data-Driven Parametrization of Molecular Mechanics Force Fields for Expansive Chemical Space Coverage」为题,发表在《Chemical Science》上。

论文链接:https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc06640e
研究背景
小分子力场是药物发现中的重要工具,在虚拟筛选,分子对接以及自由能预测等计算方法中发挥关键作用。
随着计算机辅助药物设计(AIDD)和有机合成技术的发展,药物发现中探索的化学空间范围不断扩大,迫切需求在广阔化学空间内都能够提供高精度预测的小分子力场。
近年来,量子化学和机器学习(ML)技术的发展,给上世纪发展起来的小分子力场提供了新的机遇。备受关注的机器学习力场(MLFF)尽管能够提供高精度的预测,但由于 ML 模型的复杂性,存在训练数据需求量大,推理速度慢等问题。因此很难构建一个计算效率高,且能覆盖广阔化学空间的 MLFF。
2022 年,Espaloma 力场提出了一种平衡精度和效率的方法。他们维持了传统力场的势函数形式,同时抛弃传统的查表赋参方法,使用图神经网络(GNN)预测传统力场参数。数据驱动的参数化方法能够最大限度提升传统力场的精度,同时维持了计算效率。
在此基础上,提高力场的精度与化学空间覆盖,不仅是简单的数据量放大,还需要与之匹配的模型结构和训练策略设计。
模型结构与训练策略
ByteFF 模型结构由 Featurization, GNN, Output 三层组成。
在 Featurization 层中,将每个原子和化学键的化学特征提取为向量表示。GNN 层中,使用 EGT 结构进行信息传递,充分利用原子和键的特征,获得每个原子和键的化学环境表示。Output 层中,根据结构的化学环境预测力场参数。
通过模型结构设计,ByteFF 保证了参数预测结果符合诸多物理限制,比如相同化学环境的结构有相同的结构参数预测,原子 partial charge 之和严格等于分子的总电荷等。
在训练方面,研究人员构建了含有 2.4 M 不同分子碎片的 optimization 数据集和旋转 3.2 M 不同二面角的 torsion 数据集。基于该数据集,研究者针对性地设计了 partial Hessian loss,能够 end-to-end 对批量数据进行训练。
由于传统力场形式简单,几乎不可能完美拟合量化势能面,研究者提出在 torsion 数据集上使用迭代「结构优化-训练」的策略,保证 ByteFF 能够在二面角这个关键自由度上提供准确的势能面预测。
此外,研究者们使用预训练、训练、微调等多阶段训练流程,取得了最优的训练效果。
性能评估
结构优化方面,ByteFF 显著优于业界 SOTA(OPLS4+ffbuilder,标记为「OPLS4 cst」)。
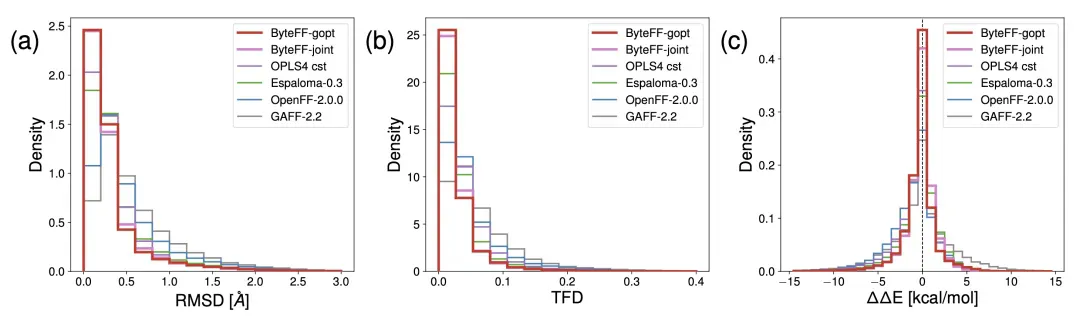
ByteFF 能够准确预测小分子包括环内和非环的二面角势能面。
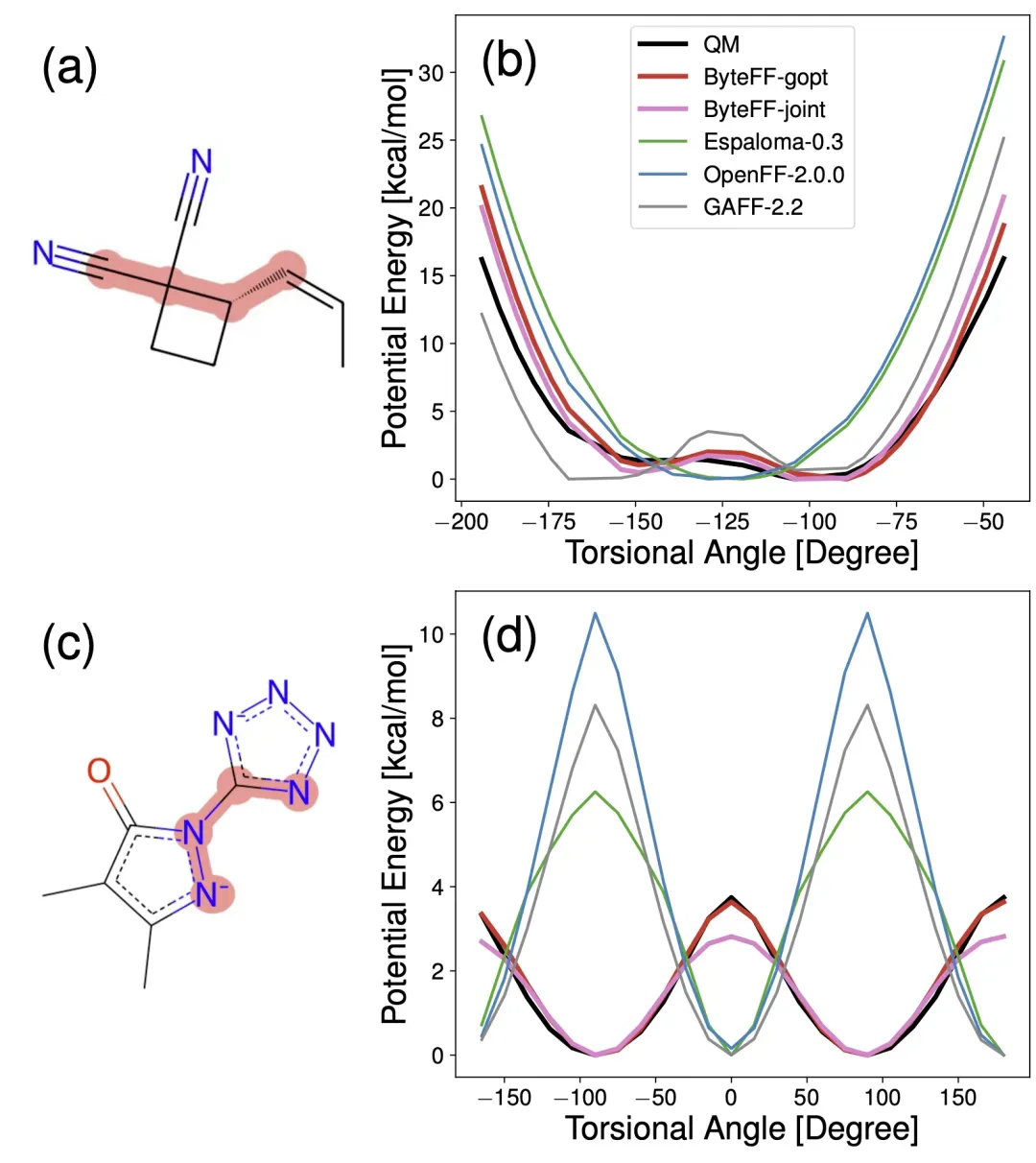
更多结果请参考原文献。
总结与展望
得益于先进的网络结构设计,充足的训练数据和与之匹配的训练流程,ByteFF 在结构优化,分子势能面预测等多个方面取得了卓越的效果。
ByteFF 继承了 GAFF2 的非键参数,虽然保证了与 Amber 力场的适配,但在非键相互作用方面仍存在较大的提升空间,是今后重要的研究方向。
目前 ByteFF 免费 API 测试正在进行中,如有需要请联系论文的通讯作者,并注明单位和用途。欢迎同行们的试用与反馈。


