
编辑 | 白菜叶
卷积神经网络(CNN)和 Transformer 等深度学习架构通过捕捉局部和长距离依赖关系,显著推进了生物序列建模。然而,它们在生物学环境中的应用受到高计算需求和对大数据集的需求的限制。
麻省理工学院、哈佛大学和卡内基梅隆大学等机构的研究人员提出了 Lyra,这是一种用于序列建模的次二次架构,它基于上位性的生物学框架,用于理解序列与功能之间的关系。
Lyra 在 100 多个广泛的生物任务中表现出色,在许多关键领域实现了 SOTA 性能,包括蛋白质适应度景观预测、生物物理特性预测(例如无序蛋白质区域功能)肽工程应用(例如抗体结合、细胞穿透肽预测)、RNA 结构分析、RNA 功能预测和 CRISPR gRNA 设计。
与当前的生物学基础模型相比,它的推理速度的大幅提升,参数大幅减少(最多减少 120,000 分之一)。
使用 Lyra,研究人员能够在不到两小时内在两个或更少的 GPU 上训练和运行生物序列建模任务。
该研究以「Lyra: An Efficient and Expressive Subquadratic Architecture for Modeling Biological Sequences」为题,于 2025 年 3 月 20 日发布在 arXiv 预印平台。

CNN 能够通过次二次缩放有效地检测局部序列模式,而 Transformers 则利用自注意力来模拟全局交互,但需要二次缩放,因此计算成本高昂。
混合模型(例如 Enformers)集成了 CNN 和 Transformers,以平衡局部和全局上下文建模,但它们仍然面临可扩展性问题。
包括 AlphaFold 和 ESM3 在内的大规模 Transformer 模型在蛋白质结构预测和序列功能建模方面取得了突破。但是,它们对大量参数缩放的依赖限制了它们在数据可用性通常受限的生物系统中的效率。这凸显了对计算效率更高的方法来准确建模序列与功能关系的需求。
为了克服这些挑战,上位效应(序列内突变之间的相互作用)为生物序列建模提供了一个结构化的数学框架。多线性多项式可以表示这些相互作用,为理解序列-功能关系提供了一种原则性方法。状态空间模型 (SSM) 自然地与此多项式结构保持一致,使用隐藏维度来近似上位效应。
与 Transformer 不同,SSM 利用快速傅里叶变换 (FFT) 卷积来高效地对全局依赖关系进行建模,同时保持次二次缩放。此外,集成门控深度卷积可通过自适应特征选择增强局部特征提取和表达能力。这种混合方法平衡了计算效率和可解释性,使其成为基于 Transformer 的生物序列建模架构的有前途的替代方案。
Lyra
来自麻省理工学院、哈佛大学和卡内基梅隆大学等机构的研究人员推出了 Lyra,这是一种专为生物应用而设计的次二次序列建模架构。Lyra 集成了 SSM 来捕获长距离依赖关系,并使用投影门控卷积进行局部特征提取,从而实现高效的 O(N log N) 扩展。

图示:Lyra 概述。(来源:论文)
它有效地对上位相互作用进行建模,并在 100 多项生物任务中实现了最先进的性能,包括蛋白质适应度预测、RNA 功能分析和 CRISPR 指南设计。Lyra 的运行参数明显更少(比现有模型小 120,000 分之一),同时推理速度提高了 64.18 倍,使高级生物序列建模变得民主化。
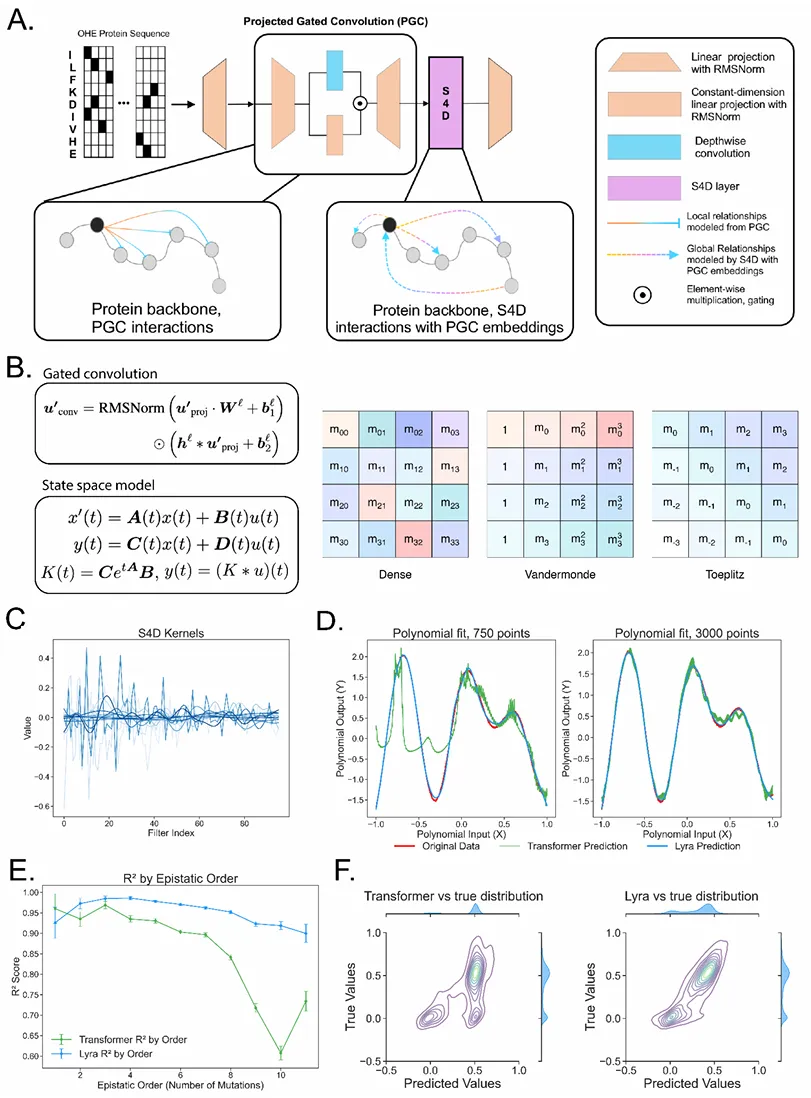
图示:Lyra 架构能够通过学习到的局部和全局关系对上位式交互进行高效建模。(来源:论文)
Lyra 由两个关键组件组成:投影门控卷积 (PGC) 块和具有深度卷积 (S4D) 的状态空间层。该模型拥有大约 55,000 个参数,包括两个用于捕获局部依赖关系的 PGC 块,后面跟着一个用于建模长距离交互的 S4D 层。
PGC 通过将输入序列投影到中间维度、应用深度 1D 卷积和线性投影以及通过元素乘法重新组合特征来处理输入序列。S4D 利用对角状态空间模型使用矩阵 A、B 和 C 计算卷积核,通过加权指数项有效捕获序列范围的依赖关系,并增强 Lyra 有效建模生物数据的能力。
Lyra 是一种序列建模架构,旨在有效捕获生物序列中的局部和长距离依赖关系。它集成了 PGC 以进行局部建模,并集成了对角化 S4D 以进行全局交互。Lyra 使用多项式表达力来近似复杂的上位性交互,在蛋白质适应度景观预测和深度突变扫描等任务中的表现优于基于 Transformer 的模型。
它在各种蛋白质和核酸建模应用中实现了 SOTA 精度,包括无序预测、突变影响分析和 RNA 依赖性 RNA 聚合酶检测,同时保持比现有大规模模型明显更少的参数数量和更低的计算成本。
结语
总之,Lyra 使用了用于生物序列建模的次二次架构,利用 SSM 有效地近似多线性多项式函数。这可以实现对上位相互作用的卓越建模,同时显著降低计算需求。

图示:Lyra 在各种蛋白质预测任务中实现了 SOTA 性能。(来源:论文)
通过集成 PGC 进行局部特征提取,Lyra 在 100 多项生物任务中实现了一流的性能,包括蛋白质适应度预测、RNA 分析和 CRISPR gRNA 设计。它的表现优于大型基础模型,参数更少,推理速度更快,仅需一到两块 GPU 即可在数小时内完成训练。
Lyra 的效率使人们能够通过治疗、病原体监测和生物制造应用获得先进的生物建模。
论文链接:https://arxiv.org/abs/2503.16351
相关内容:https://www.marktechpost.com/2025/03/24/lyra-a-computationally-efficient-subquadratic-architecture-for-biological-sequence-modeling/


