编辑 | 萝卜皮
深度生成模型在药物从头设计领域受到广泛关注。然而,针对新靶点合理设计配体分子仍然具有挑战性,特别是在控制生成分子的性质方面。
在这里,受到 DNA 编码化合物库技术的启发,湖南大学和西安电子科技大学的研究人员提出了 DeepBlock,这是一种基于块(block)的配体生成深度学习方法,可根据目标蛋白质序列进行定制,同时实现精确的属性控制。
此外,DeepBlock 还可以结合优化算法和深度学习来调控生成分子的性质。
该研究以「A deep learning approach for rational ligand generation with toxicity control via reactive building blocks」为题,于 2024 年 11 月 8 日发布在《Nature Computational Science》。

寻找能够与特定蛋白质结合的小分子配体是药物发现中的关键环节。虚拟筛选已成为一种重要方法,利用计算机程序在小分子库中识别生物活性化合物。然而,虚拟筛选的有效性受到巨大化学空间和所用化合物库的限制。
相比之下,从头药物设计策略(从头生成分子结构)提供了一种有希望的途径,可以探索现有库之外更广阔的化学空间。
近年来,这些模型在分子生成上取得了显著进展,但它们通常缺乏针对特定蛋白质靶标的生成能力,因此需要通过额外筛选或结合强化学习等技术来弥补。此外,生成分子的可合成性和药物的毒性、代谢等特性也需在实际药物开发中予以考虑。
DNA 编码化合物库技术已成为一种广泛接受的湿实验室药物发现方法。该方法利用组合化学通过分子构件的反应快速生成大量候选化合物。
DeepBlock
受 DNA 编码化合物库技术的启发,湖南大学和西安电子科技大学的研究团队提出了一种基于深度学习的框架 DeepBlock,利用分子构件进行从头药物设计。这里的构件表示能够相互发生化学反应的分子片段。
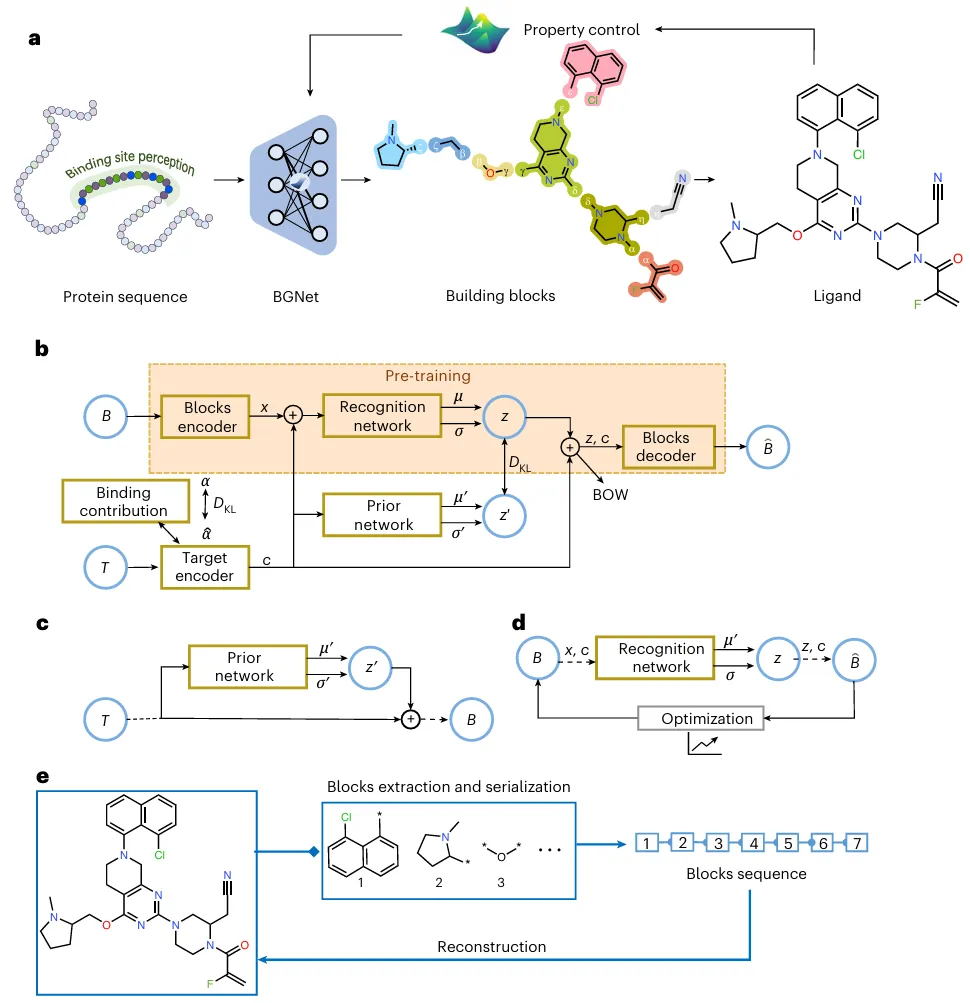
图示:DeepBlock 框架概述。(来源:论文)
DeepBlock 的核心理念是将分子生成过程分解为两个连续步骤:首先,根据蛋白质序列嵌入特征生成构建块,然后将其组装成完整的分子。通过利用这些块的固有属性及其之间的化学相互作用,DeepBlock 可以设计出更优质的合理分子。
基于这一理念,研究人员在 DeepBlock 中设计了有效的机制来解决两个关键任务:根据蛋白质序列定制分子生成和生成过程中的属性控制。
在 DeepBlock 中,该团队加入了块生成网络(BGNet),这是一种条件深度生成模型,旨在根据给定的蛋白质序列生成块序列。BGNet 结合了两个关键特性,可显著提高其性能。
首先,它由一个在大规模分子数据集上预训练的分子块自动编码器构建而成,它包含 10,701 个块的广泛词典,其中包含各种常用的片段。这种预训练扩展了化学空间,并缓解了由于蛋白质-配体对数据集的大小有限而导致的潜在过度拟合。
其次,研究人员在 DeepBlock 中引入了一个关键组件——靶标贡献感知模块。该模块增强了模型自主识别配体与残基之间相互作用的能力,弥补了蛋白质序列中 3D 结构信息的缺失。
BGNet 中这两个特征的结合凸显了其生成多样化和生物活性分子片段的能力,有效地解决了蛋白质序列数据带来的挑战。
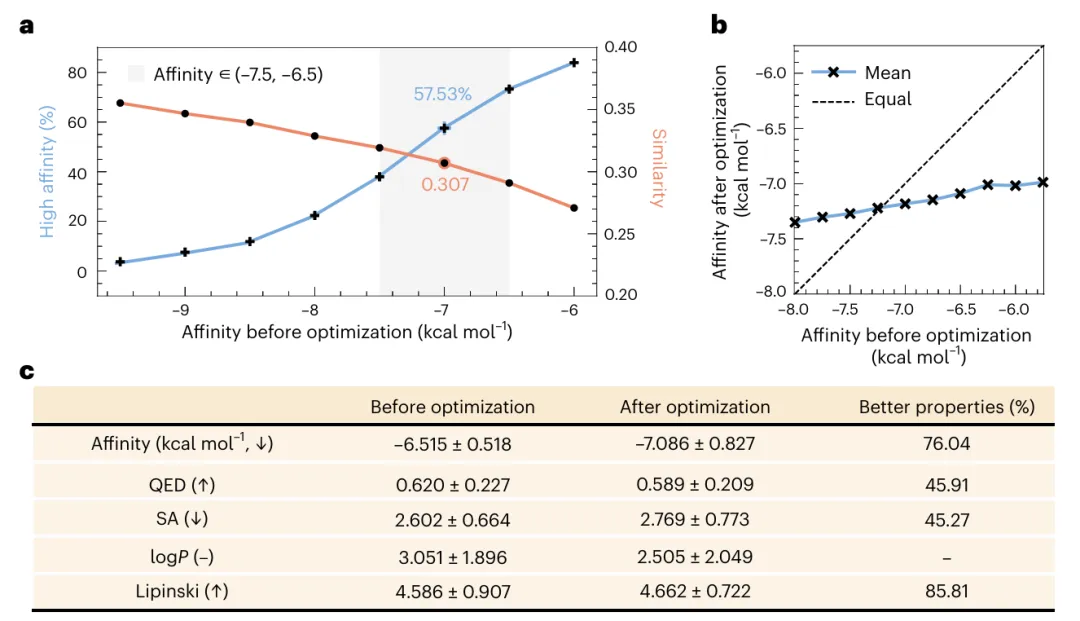
图示:优化前后亲和力对比。(来源:论文)
此外,该团队将 BGNet 与模拟退火 (SA) 算法或贝叶斯优化 (BO) 结合使用来控制生成过程,旨在增强其他特性,同时保留其对目标蛋白质的结合亲和力。
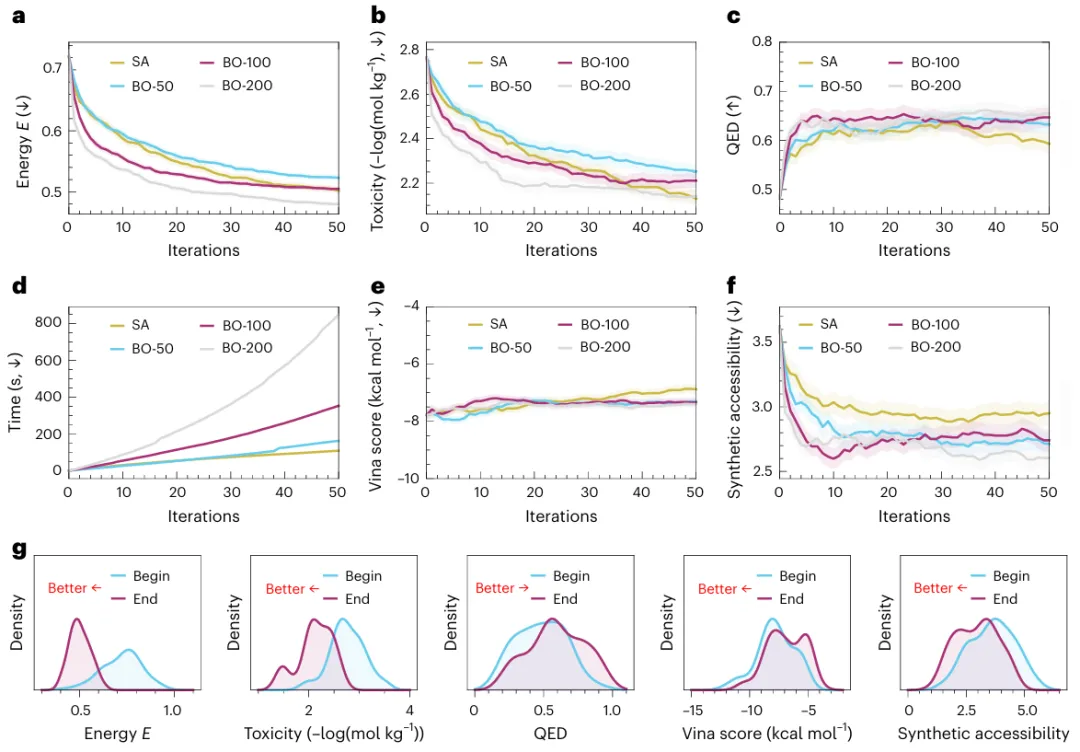
图示:优化过程和结果。(来源:论文)
研究团队还进行了以药物毒性为优化目标的实验。当与以毒性为优化目标的模拟退火或贝叶斯优化相结合时,DeepBlock 成功生成具有低毒性的配体,同时保留与靶标的亲和力。
未来工作
该方法也存在局限性。
DeepBlock 目前只能从现有的块字典中生成块,因此限制了其生成的分子的多样性。
该团队未来的研究方向是探索从头生成块的方法,从而将该模型从现有字典块的限制中解放出来,并释放其可以创建的分子的更大多功能性和新颖性的潜力。
此外,DeepBlock 生成二维 (2D) 分子结构作为 SMILES 字符串,提供可控属性和对新目标的适用性。虽然 SMILES 字符串为各种药物开发场景提供了足够的结构信息,但它们缺乏 3D 结构细节。
未来的研究将侧重于将该方法与 LiGAN 等方法相结合,以开发基于分子构建块的受控 3D 分子生成方法。这种混合方法可以结合 2D 和 3D 药物设计方法的优势,从而有可能提高药物发现的效率和有效性。
论文链接:https://www.nature.com/articles/s43588-024-00718-0