编辑 | X
传统意义上,发现所需特性的分子过程一直是由手动实验、化学家的直觉以及对机制和第一原理的理解推动的。
随着化学家越来越多地使用自动化设备和预测合成算法,自主研究设备越来越接近实现。
近日,来自 MIT 的研究人员开发了由集成机器学习工具驱动的闭环自主分子发现平台,以加速具有所需特性的分子的设计。无需手动实验即可探索化学空间并利用已知的化学结构。
在两个案例研究中,该平台尝试了 3000 多个反应,其中 1000 多个产生了预测的反应产物,提出、合成并表征了 303 种未报道的染料样分子。
该研究以《Autonomous, multiproperty-driven molecular discovery: From predictions to measurements and back》为题,于 2023 年 12 月 22 日发布在《Science》上。
论文链接:https://www.science.org/doi/10.1126/science.adi1407
发现具有所需功能特性的小分子对于健康、能源和可持续发展的进步至关重要。该过程通常是通过缓慢、费力、迭代的设计-制造-测试-分析 (DMTA) 循环进行的。
新兴的机器学习 (ML) 工具可以生成新的候选分子,预测其特性,并通过计算机辅助合成规划 (CASP) 提出反应途径。化学自动化的进步可以在手动设置后以最少的人为干预实现化学合成和表征。
将 ML 生成算法、ML 属性预测、CASP、机器人技术和自动化化学合成、纯化和表征集成到 DMTA 工作流程中,可以开发自主化学发现平台,该平台能够在不同的化学空间中运行,而无需手动重新配置。理想的以属性为中心的发现平台将提出并合成分子,以丰富机器学习生成和属性模型,并最终发现性能最佳的分子。实际上,有必要排除可用自动化硬件无法安全执行的反应。
为了实现自主发现,来自 MIT 的研究团队展示了一个集成的 DMTA 循环,该循环迭代地提出、实现和表征分子,仅在预测工具的指导下探索化学空间。
图补全(graph-completion)生成模型设计候选分子,并使用 ML 模型针对这三个属性中的每一个进行评估。CASP 工具提出了多步合成配方,由自动液体处理机、批量反应器、高性能液相色谱 (HPLC) 和机械臂执行。Plate reader 测量吸收光谱,校准的 HPLC 保留时间提供水-辛醇分配系数,模拟太阳光源与 plate reader 结合量化光氧化降解。测得的分子特性会自动反馈以重新训练特性预测模型,从而完成自动化 DMTA 循环的一个步骤。
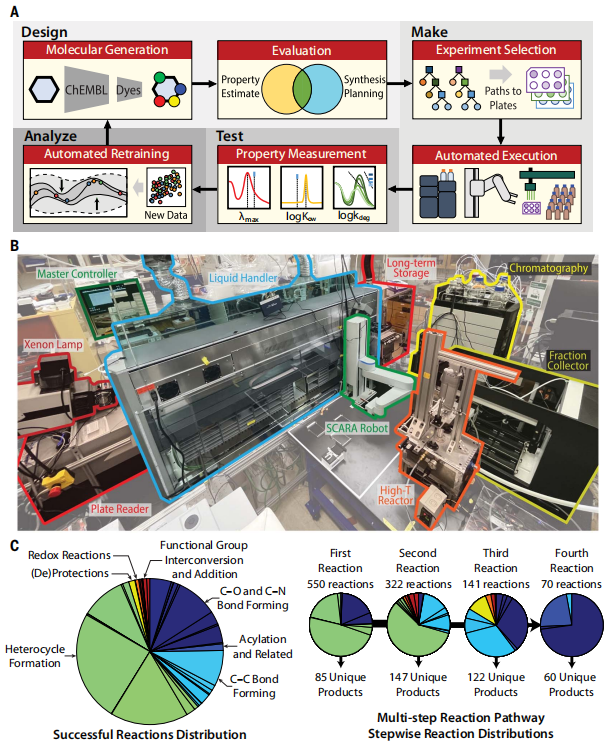
集成平台概述,以及平台预测并成功执行的反应。(来源:论文)
研究人员在小分子有机染料的两个分子发现用例中展示了该平台:(i)探索未知的化学空间和(ii)利用已知的化学空间。该平台根据需要执行并自动调整工作流程,在迭代期间和迭代之间,人工干预仅限于设定和调整目标、提供所需材料以及偶尔修复不可恢复的错误,例如 HPLC 装置堵塞。
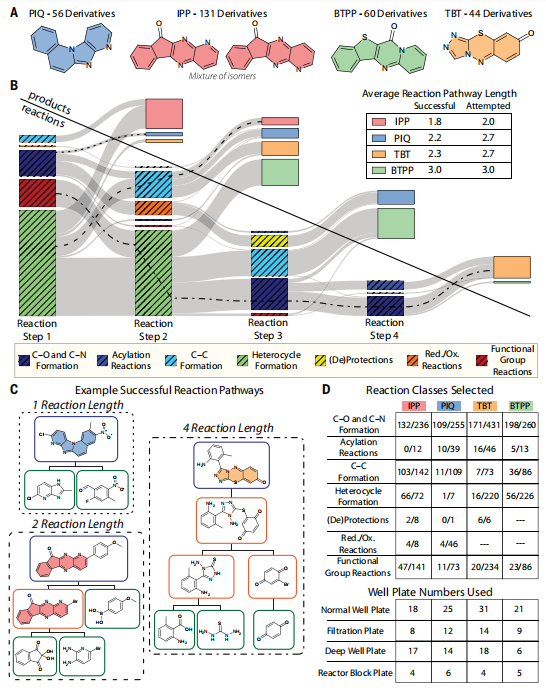
探索案例研究的化学预测和尝试。(来源:论文)
平台灵活性对于执行包含反应类别广度的多步反应途径至关重要,这些反应类别需要实现涵盖所需属性空间的候选物。研究通过针对分子染料(具有多种化学性质和复杂分子特性的模型系统)来验证该平台的功能。在两个案例研究中,该平台尝试了 3000 多个反应,其中 1000 多个产生了预测的反应产物,完成了 303 个未报告分子(即在发布之前没有 CAS 登记号)的多步反应途径。
对于这两个案例研究,吸收最大值、分配系数和光氧化稳定性是目标属性,平台自动测量和记录每个属性,以完善模型预测并为未来的实验选择提供信息。
研究人员表示:「该平台的未来迭代将受益于预测能力的改进,特别是反应保真度、条件推荐和分子生成,以及分析工具。闭环集成平台的持续开发是继续加速分子发现的一条有希望的道路。」